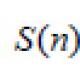Возможные кариотипы синдрома дауна. Реферат: Анализ кариотипа для определения синдромов Дауна, Клайнфельтера, Тернера
Елена Шведкина об одном из самых распространенных генетических заболеваний - больные жалуются на бесплодие, эректильную дисфункцию, гинекомастию и остеопороз
Синдром Клайнфельтера - генетическое заболевание, характеризующееся дополнительной женской половой хромосомой Х (одной или даже несколькими) в мужском кариотипе ХY . При этом в мужских половых железах - яичках - образуется недостаточно половых гормонов.
Как известно, генетический набор человека насчитывает 46 хромосом, из которых 22 пары называются соматическими, а 23‑я пара - половая. Женщины имеют пару половых хромосом ХХ , а мужчины - ХY . Для синдрома Клайнфельтера обязательно наличие мужской Y-хромосомы, поэтому, несмотря на дополнительные Х -хромосомы, пациенты всегда являются мужчинами.
Классификация: виды кариотипов при синдроме Клайнфельтера
По количеству дополнительных Х-хромосом различают следующие варианты синдрома Клайнфельтера:
- 47,ХХY - наиболее часто встречающийся
- 48,ХХХY
- 49,ХХХХY
Кроме того, к синдрому Клайнфельтера также относят мужские кариотипы, включающие, помимо дополнительных Х -хромосом, дополнительную Y -хромосому - 48,ХХYY . И, наконец, среди пациентов с этим синдромом встречаются лица с мозаичным кариотипом 46,ХY /47,ХХY (то есть часть клеток имеет нормальный хромосомный набор).
История открытия синдрома
Синдром получил свое название в честь Гарри Клайнфельтера - врача, в 1942 году впервые описавшего клиническую картину болезни. Клайнфельтер с коллегами опубликовали отчет об обследовании 9 мужчин, объединенных общими симптомами, такими как слабое оволосение тела, евнухоидный тип телосложения, высокий рост и уменьшенные в размерах яички. Позднее, в 1956 г., генетики Планкетт и Барр (Е. R. Plankett, М. L. Barr) обнаружили у мужчин с синдромом Клайнфельтера тельца полового хроматина в ядрах клеток слизистой оболочки полости рта, а в 1959 году Полани и Форд (P. E. Polanyi, S. E. Ford) с сотрудниками показали, что у больных в хромосомном наборе имеется лишняя Х -хромосома.
Активные исследования данной патологии велись в 70‑х годах в США. Тогда всех новорожденных мальчиков подвергали кариотипированию, в результате чего удалось достоверно выявить распространенность и генетические особенности синдрома Клайнфельтера.
Любопытно, что мыши также могут иметь синдром трисомии по половым хромосомам XXY, что позволяет эффективно использовать их в качестве моделей для исследования синдрома Клайнфельтера.
Распространенность заболевания
Синдром Клайнфельтера является одним из наиболее распространенных генетических заболеваний: на каждые 500 новорождённых мальчиков приходится 1 ребёнок с данной патологией.
Кроме того, синдром Клайнфельтера - третья по распространенности эндокринная патология у мужчин (после сахарного диабета и патологии щитовидной железы) и наиболее частая причина врожденного нарушения репродуктивной функции у мужчин.
На сегодняшний день около половины случаев синдрома Клайнфельтера остаются нераспознанными. Часто такие пациенты обращаются за помощью по поводу бесплодия, эректильной дисфункции, гинекомастии, остеопороза, анемии и пр. без установленного ранее диагноза.
Этиология и причины нарушения
Синдром Клайнфельтера относится к генетическим заболеваниям, не передающимся по наследству, поскольку больные, за редким исключением, бесплодны. Патология, как правило, возникает в результате нарушения расхождения хромосом на ранних стадиях формирования яйцеклеток и сперматозоидов. При этом синдром Клайнфельтера, возникающий за счет нарушения в женских половых клетках, встречается в три раза чаще. Мозаичные формы обусловлены патологией деления клеток на ранних стадиях эмбриогенеза, поэтому часть клеток у таких пациентов имеет нормальный кариотип. Причины нерасхождения половых хромосом и нарушения деления клеток на самых ранних стадиях эмбриогенеза до сих пор малоизучены. В отличие от других хромосомных заболеваний, влияние возраста родителей отсутствует или выражено незначительно.
Ранние признаки
В отличие от большинства заболеваний, связанных с нарушением количества хромосом, внутриутробное развитие детей с синдромом Клайнфельтера проходит нормально, склонности к преждевременному прерыванию беременности не наблюдается. Так что в младенческом и раннем детском возрасте заподозрить патологию практически невозможно. Более того, клинические признаки классического синдрома Клайнфельтера проявляются, как правило, только в подростковом периоде. Однако есть симптомы, которые позволяют заподозрить наличие синдрома Клайнфельтера в препубертатном периоде:
- высокий рост (пик прибавки роста приходится на период между 5–8 годами);
- длинные ноги (непропорциональное телосложение);
- высокая талия.
У части пациентов наблюдается некоторая задержка в развитии речи.
В подростковом возрасте синдром часто проявляется гинекомастией, которая при данной патологии имеет вид двустороннего симметричного безболезненного увеличения грудных желез. Так как такого рода гинекомастия часто наблюдается у совершенно здоровых подростков, этот симптом часто остается без внимания. В норме подростковая гинекомастия бесследно исчезает в течение нескольких лет, у пациентов же с синдромом Клайнфельтера обратной инволюции грудных желез не происходит. В некоторых случаях гинекомастия может не развиваться вовсе, и тогда патология проявляется признаками андрогенной недостаточности уже в постпубертатный период.
Симптомы андрогенной недостаточности при синдроме Клайнфельтера
Андрогенная недостаточность при синдроме Клайнфельтера связана с постепенной атрофией яичек, что приводит к снижению синтеза тестостерона. Степень недостаточности андрогенов резко варьирует.
В первую очередь обращают на себя внимание внешние признаки гипогонадизма:
- скудная растительность на лице или же полное ее отсутствие;
- рост волос на лобке по женскому типу;
- волосы на груди и других частях тела отсутствуют;
- маленький объем яичек (2–4 мл) и их плотная консистенция (патогномоничный признак).
Поскольку дегенерация половых желез, как правило, развивается в постпубертатный период, у большинства пациентов размеры мужских половых органов, за исключением яичек, соответствуют возрастным нормам.
Пациенты могут жаловаться на ослабление либидо и снижение потенции. У многих мужчин с синдромом Клайнфельтера половое влечение вовсе не возникает, а некоторые - напротив, заводят семью и живут нормальной половой жизнью. Наиболее постоянный признак патологии - бесплодие, именно оно чаще всего становится причиной обращения таких пациентов к врачу. У 10 % мужчин с азооспемией обнаруживают синдром Клайнфельтера.
Всем пациентам с нарушениями сперматогенеза необходимо определять кариотип для исключения или подтверждения диагноза синдрома Клайнфельтера.
Недостаток андрогенов приводит к развитию остеопороза, анемии и слабости скелетной мускулатуры. У трети больных можно наблюдать варикозное расширение вен голеней.
Андрогены влияют на обмен веществ, поэтому больные с синдромом Клайнфельтера склонны к ожирению, нарушению толерантности к глюкозе и сахарному диабету второго типа.
Доказана предрасположенность таких пациентов к аутоиммунным заболеваниям (ревматоидный артрит, системная красная волчанка, аутоиммунные заболевания щитовидной железы и другие).
Психологические особенности
Коэффициент интеллекта у больных с классическим синдромом Клайнфельтера варьирует от значений ниже среднего до показателей, значительно превышающих средний уровень. Однако во всех случаях отмечается диспропорция между общим уровнем интеллекта и вербальными способностями, так что нередко пациенты с достаточно высоким IQ испытывают трудности при восприятии больших объемов материала на слух, а также при построении фраз, содержащих сложные грамматические конструкции. Такие особенности причиняют пациентам много неприятностей в период обучения и нередко продолжают сказываться на профессиональной деятельности.
Данные о психологических особенностях больных с синдромом Клайнфельтера достаточно противоречивы, однако большинство специалистов оценивают пациентов как скромных, робких людей с несколько заниженной самооценкой и повышенной чувствительностью. Есть данные, свидетельствующие о склонности пациентов с синдромом Клайнфельтера к гомосексуализму, алкоголизму и наркомании. Сложно сказать, вызваны ли особенности психики у таких больных непосредственным влиянием хромосомной аномалии, или же это реакция на проблемы в сексуальной сфере.
В отношении разных цитогенетических вариантов синдрома Клайнфельтера справедливо правило, что с увеличением количества дополнительных Х -хромосом увеличивается количество и выраженность патологических симптомов.
Диагностика синдрома Клайнфельтера
Во многих странах синдром Клайнфельтера часто диагностируется ещё до рождения ребёнка, так как многие женщины позднего детородного возраста, в связи с высоким риском генетических дефектов у будущего потомства, используют пренатальную генетическую диагностику плода. Нередко пренатальное выявление синдрома Клайнфельтера является поводом для прерывания беременности, в том числе и по рекомендации врачей. В России анализ кариотипа будущего ребёнка проводится крайне редко.
При подозрении на синдром Клайнфельтера проводят лабораторный анализ крови для определения уровня мужских половых гормонов. Необходима дифференциальная диагностика с другими заболеваниями, протекающими с проявлениями андрогенной недостаточности. Точный диагноз синдрома Клайнфельтера ставят на основании изучения кариотипа (набора хромосом) больного.
Исследования, необходимые для подтверждения диагноза
У всех мужчин с резко повышенными концентрациями гонадотропинов необходимо исключить синдром Клайнфельтера, так как нередко первый лабораторный признак этой генетической патологии - повышение в крови концентрации гонадотропинов при нормальном содержании общего тестостерона.
Синдром Клайнфельтера необходимо дифференцировать от других форм первичного гипогонадизма. В любом случае при повышении уровня ФСГ в крови необходимо определение кариотипа для исключения в первую очередь синдрома Клайнфельтера.
Лечение
Цели лечения синдрома Клайнфельтера:
- Восстановление нормального содержания тестостерона
- Восстановление сексуальной функции
- Ликвидация метаболических нарушений
При клинически выраженной патологии необходима пожизненная заместительная терапия препаратами тестостерона. Адекватная терапия позволяет не только улучшить внешний вид и общее самочувствие больного, но и вернуть способность к нормальной половой жизни. Кроме того, заместительная терапия предупреждает развитие остеопороза, купирует мышечную слабость. В юном возрасте лечение необходимо начинать сразу же после постановки диагноза. При синдроме Клайнфельтера лучше использовать препараты тестостерона длительного действия:
- смесь эфиров тестостерона в виде масляного раствора, инъекции которого необходимо делать 2–3 раза в месяц;
- тестостерона ундеканоат в виде масляного раствора - препарат-депо с замедленным высвобождением действующего вещества - инъекции 1 раз в 3 месяца.
Гормонолечение при наличии Х хромосомы у мужчин должно носить постоянный характер. Дозу препарата подбирают индивидуально под контролем уровня тестостерона и ЛГ в сыворотке крови.
Уже развившаяся гинекомастия при синдроме Клайнфельтера не подвергается инволюции даже в случае адекватного лечения, поэтому часто приходится прибегать к хирургической коррекции (мастэктомии).
Для профилактики таких сопутствующих заболеваний, как ожирение и сахарный диабет второго типа, больным рекомендуют придерживаться диеты и следить за собственным весом.
Мониторинг пациентов с синдромом Клайнфельтера следует осуществлять не реже 1 раза в 6–12 месяцев. Он должен включать следующие исследования:
- общий анализ крови для оценки уровня гемоглобина и гематокрита;
- гормональный анализ крови, включающий определение тестостерона и ЛГ (проводится на фоне лекарственной терапии за 1–2 дня до очередной инъекции тестостерона);
Кариотип - (от карио. греч. káryon - орех, ядро и греч. týpos - образец, форма, тип) набор хромосом, совокупность признаков хромосом (количество, размеры, форма) в клетках тела организма того или иного вида. Исследование проводят в период метафазы клеточного деления. Частой причиной генетического бесплодия/невынашивания является изменение количества хромосом или изменение их структуры. Поэтому исследование кариотипа показано (при бесплодии) обоим супругам. Хромосомы представляют собой молекулы ДНК упакованные вместе с белками, необходимыми для обеспечения функционирования ДНК. В ядре всех соматических клеток человека находится 46 хромосом. Из 46 хромосом 44 или 22 пары - аутосомные хромосомы, последняя пара - половые хромосомы. У женщин половые хромосомы в норме представлены двумя Х-хромосомами, у мужчин - двумя хромосомами Х и Y. Во всех парах хромосом как аутосомных, так и половых одна из хромосом получена от отца, а вторая от матери. В половых клетках - в сперматозоиде и в яйцеклетке содержится 23 хромосомы (гаплоидный набор). Сперматозоиды делятся на два типа в зависимости от того, содержат они хромосому Х или Y. яйцеклетки в норме содержат только хромосому Х. В хромосомах сосредоточено около 99% всей ДНК клетки, оставшаяся часть ДНК находится в других клеточных органеллах (например, в митохондриях). ДНК в хромосомах эукариот находится в комплексе с основными белками - гистонами и с негистоновыми белками, которые обеспечивают сложную упаковку ДНК в хромосомах и регуляцию её способности к синтезу рибонуклеиновых кислот (РНК). Ежегодно в литературе появляется большое количество описаний новых генетически детерминированных аномалий. По одним из данных известно более 2000 наследственных синдромов у человека. По статистике около 0,7% детей рождаются с множественными пороками развития. Нарушения кариотипа часто сопровождаются пороками развития, которые несовместимы с жизнью, что заканчивается внутриутробной гибелью плода и абортом. Однако, часть дефектов в кариотипе позволяют донашивать плод и ребенок рождается с присущими фенотипическими и генотипическими признаками для той или иной болезни или синдрома. К основным аномалиям кариотипа можно отнести: синдром Дауна, синдром Шерешевского- Тернера, синдром Эдвардса, синдром Клайнфельтера. Хромосомные нарушения выявляются по меньшей мере у 10% оплодотворенных яйцеклеток и у 5-6% плодов. Самопроизвольный аборт при хромосомных дефектах регистрируется обычно на 8-11-й неделе беременности (возможны более поздние самопроизвольные аборты и мертворождения). По результатам обследований 65000 новорожденных, проведенных в разных лабораториях, значительные хромосомные аберрации или изменение числа хромосом выявляются примерно у 0,5% детей. По крайней мере у 1 из 700 детей имеется трисомия по 21, 18 или 13-й хромосоме; приблизительно у 1 из 350 новорожденных мальчиков - кариотип 47,XXY или 47,XYY ; у одного ребенка на каждые несколько тысяч новорожденных - моносомия по Х-хромосоме; у одного из 500 - хромосомные аберрации, большинство которых генетически компенсированы. При обследовании взрослых изредка выявляют наследуемые компенсированные хромосомные аберрации, а также некоторое число людей с кариотипами 47,XXY , 47,XYY и 47,XXX . При умственной отсталости хромосомные аномалии находят у 10-15% больных, и еще чаще при сопутствующих анатомических дефектах. Мужчины, страдающие бесплодием или поведенческими расстройствами, часто имеют лишнию Х- или Y-хромосому. У женщин при бесплодии и пониженной фертильности часто обнаруживают аберрации Х-хромосомы или моносомию по Х-хромосоме. При первичной аменорее аберрации Х-хромосомы находят примерно у четверти женщин. Хромосомные аберрации нередко обнаруживают при бесплодии как у мужчин, так и у женщин. К наиболее распространенным хромосомным мутациям относят трисомии. Трисомией называют появление в кариотипе дополнительной хромосомы. Наиболее известные примеры - болезнь Дауна, которую также называют трисомией по хромосоме 21. Трисомия по хромосоме 13 - синдром Патау и по хромосоме 18 - синдром Эдвардса. Эти трисомии являются аутосомными. Другие трисомики по аутосомам нежизнеспособны, погибают внутриутробно. Жизнеспособными являются индивидуумы с дополнительными половыми хромосомами. Трисомия по половым хромосомам может быть трех типов - 47,XXY; 47,XXХ; 47,XYY (трисомия 47,XXY, известна как синдром Клайнфельтера). Клинические проявления дополнительных хромосом Х или Y могут быть и незначительными. Трисомии 47,ХХY и 47,ХYY встречаются с частотой 1:1000 среди женщин и мужчин соответственно, проявляются относительно небольшими фенотипическими проявлениями и обычно обнаруживаются как случайные находки. Синдром Дауна (синонимы: трисомия по 21-й хромосоме, G 1 -трисомия). Описан Down JLH в 1866 году. Одна из самых частых врожденных болезней человека (1 на 660 новорожденных по данным Penrose L.S.,Smith G.F.1966). Отличительные признаки - умственная отсталость, мышечная гипотония, плоское лицо, монголоидный разрез глаз, маленькие ушные раковины. Вероятность нерасхождения хромосом в женских половых клетках увеличивается с возрастом матери. Частота рождения больного ребенка у женщин 15-29 лет составляет 1 на 1500 родов, 30-34 лет - 1 на 800, 35-39 лет 1 на 270, 40-44 лет 1 на 100, после 45 лет 1 на 50. Синдром Дауна вызван трисомией по всей 21-й хромосоме или по ее большей части. Исходя из обобщенных данных исследований относительная частота хромосомных аберраций для этого синдрома следующая: 1. Полная трисомия по 21-й хромосоме - 94%; 2. Мозаицизм, сочетающий трисомию с нормальным набором хромосом - 2,4%; 3. Транслокация 21-й хромосомы или ее большей части на хромосомы группы D или G (примерно с одинаковой частотой) - 3,3%. Мозаицизм вызывает менее тяжелые проявления, умственное развитие задерживается, а может и не нарушаться, что нельзя предсказать по внешнему виду. Мозаицизм - существование в организме двух или более генетически разных типов клеток. Хорошо развитые дети с внешностью характерной для синдрома Дауна, скорее всего имеют мозаицизм, что иногда не легко подтвердить. Средний IQ у больных подростков и взрослых (по некоторым оценкам) равен 24. По статистике в 1983 году больные с синдромом Дауна с среднем доживали до 25 лет, а в 1997 уже до 49 лет. К основной причиной ранней смерти относят врожденные пороки сердца, а также болезни органов дыхания, лейкозы. Наблюдается ослабление гуморального и клеточного иммунитета. Из сопутствующих заболеваний наиболее распространены трудно поддающиеся лечению ринит, конъюктивит, пародонтит. Синдром Эдвардса (синонимы: трисомия по 18-й хромосоме, Е 1 - трисомия). Впервые описан Edwards JH в 1960 году. Второй по частоте синдром множественных пороков развития. Встречается с частотой 1 на 3000 новорожденных (больных девочек рождается в 3 раза больше, чем мальчиков). Описано более 130 симптомов этой хромосомной аномалии. Отличительные признаки - сжатые кулаки с перекрытием пальцев, короткая грудина, кожный узор в виде дуг на большинстве пальцев. Синдром Эдвардса вызван трисомией по 18-й хромосоме или по ее крупной части. У большинства пациентов обнаруживается полная трисомия, из-за неправильного расхождения хромосом в мейозе. Вероятность такого расхождения увеличивается с возрастом матери. Мозаичная форма трисомии по 18-й хромосоме протекает легче полной трисомии. Фенотип колеблется от почти нормального до развернутой формы заболевания. Частичная форма проявляется по-разному - в зависимости от того, какая часть хромосомы удвоена. Трисомия по короткому плечу сопровождается смазанной клинической картиной с нормальным психическим развитием ил легкой умственной отсталостью. Дети с данным синдромом рождаются слабыми, половина детей погибают на первой недели жизни, немногие доживают до года. средняя продолжительность жизни- 14,5 дня, дети переживающие год (5-10%) страдают глубокой умственной отсталостью. Известны единичные случаи выживания детей свыше 10 лет. Синдром Патау (синонимы: трисомия по 13-й хромосоме, D 1 - трисомия). Впервые описан Patau K в 1960 году. Встречается с частотой 1 на 5000 новорожденных. Отличительные признаки - пороки развития глаз, носа и верхней губы, прозэнцефалические пороки, полидактилия, длинные выпуклые ногти, очаговая аплазия кожи волосистой части головы. Синдром вызван трисомией по 13-й хромосоме или по ее крупной части. Мозаичная форма трисомии протекает, как правило, легче с различной выраженностью симптомов и степенью умственной отсталости. Продолжительность жизни выше. Трисомия по короткому плечу и проксимальной части длинного плеча 13-й хромосомы проявляется неспецифическими признаками и обычно выраженной умственной отсталостью. Трисомия по дистальной части хромосомы проявляется глубокой умственной отсталостью и гибелью в раннем неонатальном периоде. Половина детей умирают на первой недели после рождения и только один из десяти переживает один год. Синдром Тернера (синонимы: сексогенная карликовость, синдром ХО, синдром моносомии по Х-хромосоме, синдром Улльриха, синдром Шерешевского-Тернера). Подробно описан Turner HH в 1938году. Впервые замечен Rossle RI в 1922 году. Встречается с частотой 1 на 2500 новорожденных девочек. Отличительные признаки - низкорослость, широкая грудная клетка, гипертелоризм сосков, врожденный лимфатический отек кистей и стоп. Причиной синдрома является нерасхождение хромосом в мейозе с образованием кариотипа 45,ХО. Отсутствует полностью или частично одна из двух Х хромосом. Чаще отсутствует отцовская хромосома. Наиболее частые проявления болезни - низкорослость и дисгенезия гонад (недоразвитие или полное отсутствие фолликулов, атрофия яичников). Поскольку дисгенезия не проявляет себя до полового созревания, то девочкам с задержкой роста при отсутствии симптомов, исключающих синдром Тернера можно рекомендовать поведение цитогенетическое обследование. Мозаичная форма болезни - кариотип 46,ХХ/45,ХО или 46,ХY/45Х и неполная моносомия по Х-хромосоме (изохромосома Х или делеция части Х-хромосомы) часто протекает в легком варианте. Цитогенетическое исследование целесообразно проводить всем девочкам у которых к 13 годам отсутствуют телархе и адренархе, а также имеется первичная или вторичная аменорея с повышенным содержанием ФСГ. Показано, что в период внутриутробного развития яичники развиваются нормально, однако, примордиальных фолликулов, по-видимому, не образуется и яичники в дальнейшем атрофируются. Задержка роста у девочек иногда заметна при рождении. До з-х лет ребенок растет нормально, но с запаздыванием созревания костной ткани, а с 3-х до 12 лет, наоборот, костная ткань созревает нормально, но рост замедлен. После 12 лет замедлен рост и созревание костей, появляется склонность к полноте. Рост без лечения составляет (в среднем) 143 см. Из-за развивающейся атрофии яичников такие женщины бесплодны. У взрослых с синдромом Тернера повышена частота расслоения аорты. Повышена заболеваемость артериальной гипертензией, сахарным диабетом, артериальной гипертонией, инсультом. У 6% девочек присутствует мозаичный кариотип - 45,ХО/46,ХY и у них значительно повышен риск появления гонадобластомы. Синдром Клайнфельтера (синонимы: синдром XXY, синдром 47, XXY, синдром Клайнфельтера-Рейфенщтейна-Олбрайта). Описан Klinefelter HF в 1942году. Встречается с частотой 1 на 500 новорожденных мальчиков. Отличительные признаки: гипогонадизм, длинные ноги, снижение интеллекта, поведенческие расстройства. Проявление синдрома связано с наличием добавочной Х-хромосомы в мужском кариотипе. Причиной, примерно в половине случаев является нерасхождение хромосом в 1-м делении мейоза во время сперматогенеза, другая половина - нарушение оогенеза, и в небольшом количестве случаев - нарушение митоза в оплодотворенных клетках. Чем старше становится мужчина, тем чаще у него обнаруживаются сперматозоиды с обеими половыми хромосомами, т.е. риск рождения ребенка с синдромом Клайнфельтера должен быть выше. Синдром является наиболее частой причиной мужского гипогонадизма и бесплодия. Для таких пациентов с детства характерно евнухоидное телосложение - высокий рост, непропорционально длинные конечности, длинные ноги. Речевое развитие запаздывает, проявляется психический инфантилизм, неуверенность, или наоборот, самоуверенность, нарушенное суждение. Половой член и яички относительно невелики уже с детства, синтез тестостерона, за редким исключением, снижен вдвое. Вторичные признаки развиты слабо, у трети подростков гинекомастия. К редким симптома при синдроме Клайнфельтера относят: крипторхизм, сколиоз, сахарный диабет, хронический бронхит, легкая атаксия, трофические язвы голеней, варикозное расширение вен, тромбоз глубоких вен, остеопороз, рак молочной железы (в 20 раз чаще), экстрагонадные опухоли (чаще в возрасте 15-30 лет), аутоиммунные заболевания. В детстве симптоматика минимальна, клиническая картина развивается в пубертантный и постпубертантный период и отражает степень недостатка андрогенов. При мозаичной форме синдрома (46,XY/47,XХY) заболевание протекает легче с меньшими нарушениями яичек. Вариант синдрома Клайнфельтера - синдром XXYY характеризуется более тяжелой умственной отсталостью и выраженными поведенческими расстройствами. Синдромы ХХХ и ХХХХ (синонимы: полисомии по Х-хромосоме, синдром ХХХ - синдром трипло-Х, синдром трисомии по Х-хромосоме, синдром ХХХХ - синдром тетрасомии по Х-хромосоме, синдром тетра-Х). Синдром ХХХ описан Jacobs PA с соавт. в 1959 году. Кариотип 47,ХХХ встречается с частотой 1 на 1000 новорожденных девочек. Проявление синдрома связано с наличием добавочной Х-хромосомы (одной или двух) в женском кариотипе. Причиной синдрома ХХХ, в основном, является нерасхождение хромосом во время 1-го деления мейоза. У таких больных часто нарушена моторная речь, ослаблена слуховая память, приобретение двигательных навыков происходит с запаздыванием, типичны плохая координация движений и неуклюжесть. IQ снижено (80 -90). У трети подростков отмечаются поведенческие расстройства - замкнутость, антисоциальное поведение, легкая депрессия. Со временем эти нарушения проходят. Половое созревание происходит нормально. Синдром ХХХХ описан Carr DH с соавт. в 1961 году. Для этого синдрома из клинических проявлений характерна умственная отсталость. Рост нормальный или высокий. Черты лица напоминают синдром Дауна. IQ снижено (в среднем 55). Характерными является задержка развития речи и поведения. У таких больных часты нарушения менструального цикла и снижена фертильность, но их дети обычно здоровы. Синдром ХХХХХ (синонимы: синдром пентасомии по Х-хромосоме, синдром пента-Х). Синдром ХХХХХ описан Kesaree N и Wooley PV в 1963 году. Отличительные признаки: монголоидный разрез глаз, открытый артериальный разрез глаз, маленькие ладони, клинодикталия пятого пальца рук. Синдром обусловлен наличием трех дополнительных Х-хромосом в кариотипе женщин. Дополнительные хромосомы достаются от матери. Для этого синдрома из клинических проявлений характерны умственная отсталость, задержка роста, низкий рост, микроцефалия, слегка монголоидный разрез глаз, запавшая переносица, короткая шея, низкая линия роста волос, неправильный прикус, врожденные пороки сердца - открытый митральный порок, дефект межжелудочковой перегородки. IQ в пределах 20-75. Синдром кошачьего глаза (синонимы: синдром коломбы радужки и атрезии заднего прохода, синдром Шмида-Фраккаро). Отличительные признаки: коломба радужки, антимонголоидный разрез глаз, атрезия заднего прохода. У таких больных обнаруживают лишнюю хромосому, состоящую из двух одинаковых участков 22-й хромосомы, содержащих все короткое плечо вместе со спутниками, центромеру и короткую часть длинного плеча. Т.е. этот участок присутствует в 4-х копиях. Иногда болезнь обусловлена удвоением сегмента 22q11. Коломба радужки и атрезия заднего прохода, как главные симптомы болезни, присутствуют одновременно лишь в 9% случаев. Для заболевания характерны: легкая умственная отсталость, иногда задержка эмоционального развития при нормальном интеллекте, небольшой гипертелоризм глаз, нижняя коломба радужки или сетчатки, антимонголоидный разрез глаз, предушные ямки, ушные привески, врожденные пороки сердца более чем у трети больных (полное аномальное впадение легочных вен, дефекты межпредсердной и межжелудочковой перегородки), атрезия заднего прохода в сочетании с прямокишечными свищами, гипоспадия, гидронефроз, агенезия почек, пузырно-мочеточниковый рефлюкс. К редким симптомам относят: микроцефалию, тугоухость, стеноз наружного слухового прохода, атрезию желчных протоков, расщелину неба, поликистоз почек, дивертикул Меккеля и другие. Синдром трисомии по 8-й хромосоме. Первые работы по описанию синдрома относятся к 1963 году. Синдром обусловлен трисомией по 8-й хромосоме, как правило, это мозаичная трисомия, полная трисомия, по-видимому, редко совместима с жизнью. Для этого синдрома из клинических проявлений характерны: умственная отсталость различной степени тяжести, длинное узкое туловище, рост от низкого до высокого, аномалии лопаток и грудины, короткая шея, узкий таз, дисплазия тазобедренных суставов, пороки развития сердца, почек, мочеточников, плохая координация движений, выпуклый лоб, глубоко посаженные глаза, широкая переносица, широкие ноздри, пухлые губы, вывернутая нижняя губа, нижняя микрогнатия, узкое высокое небо/расщелина неба, большие чашеобразные уши с толстым завитком, камптодактилия 2-5 пальцев рук и ног, неполная супинация в локтевом суставе, глубокие ладонные и подошвенные борозды, контрактуры крупных суставов, аномальные ногти. К редким симптомам относят: аплазия надколенников, раздвоенные волосы, кондуктивная тугоухость, аномальное строение позвонков (расщепление позвонков, добавочный поясничный позвонок), сколиоз, крипторхизм, удвоение тощей кишки, агенезия мозолистого тела, герминогенные опухоли, лейомиосаркома желудка. Прогноз заболевания определяется тяжестью умственной отсталости.
Вопрос по анализу на кариотип: болезнь или синдром дауна?
уля2 : Подробно вопрос о кариотипе и типах синдрома Дауна рассматривается и обсуждается здесь: http://downsyndrome.borda.ru/?1-1-40-00000031-000-0-0-1230646216 Модератор не знаю где можно задать этот вопрос решила здесь,можно ли определить по анализу на кариотип болезнь или синдром дауна?наш кардиолог предложила оформить инвалидность из-за большого порока сердца,добавив при этом «тем более у вас синдром дауна»когда я сказала что при синдроме инвалидность не дают,ответила сделаете анализ узнаете болезнь у вас или синдром. С анализом на кариотип у нас заморочка вышла, делали 35дней,обьяснили что вначале роста не было,а потом выросло.Я не знаю как в генетике,но по моему если и должно что то вырасти,то сразу, тем более из форума узнала что анализ делается от4дней до двух недель,может я не права?
Meri : Слово «синдром» означает набор признаков или характерных черт. Синдром Дауна, как показал в 1959 году французский ученый Жером Лежен (Lejeune), — это генетическое состояние, которое существует с момента зачатия и определяется наличием в клетках человека дополнительной хромосомы. Свое название это состояние получило по имени английского врача Джона Лэнгдона Дауна (Down), впервые описавшего его признаки в 1866 году. Тело человека состоит из миллионов клеток, каждая из которых обычно содержит 46 хромосом. Хромосомы расположены парами — половина от матери, половина от отца. У людей с синдромом Дауна в 21-й паре присутствует дополнительная хромосома, вследствие чего, в клетках оказывается по 47 хромосом. При этом у родителей, как правило, нормальный генотип. click here Болезнь дауна есть только в головах некомпетентных врачей. click here Почитайте здесь, еще где-то обсуждалась подобная тема, но уже нелегко найти.
уля2 : Meri пишет: Болезнь дауна есть только в головах некомпетентных врачей. я тоже так считала но после разговора с врачем начала сомневаться,особенно после ее слов»я же врач,мне виднее»и теперь не знаю стоит ли нам пересдавать анализ чтобы узнать наверняка,синдром у нас или болезнь.
Надежда : Как говорили мне врачи-генетики, раньше, до повальной проверки кариотипов синдром писали при наличии просто похожих черт (внешне, физиологические, подходящие к синдрому), а если подтверждалось анализом кариотипа — ставили болезнь. Градация существует только по формам — полная трисомная, трансгенная и мозаичная. Хотя наши чудо-врачи еще кроме синдрома и болезни могут и «даунизм» выделить Девочки на форуме писали, что даже неврологи этим грешат. Так что любая из мам, которая столкнулась с этой проблемой, боюсь, знает намного больше любого врача.
G-NA85 : Надежда пишет: Хотя наши чудо-врачи еще кроме синдрома и болезни могут и «даунизм» выделить Да, Аполлинарии в карточке невропвтолог написал. Я заставила его на Синдром Дауна переделать. Так красивее звучит. Болезни Дауна — нет. Есть Синдром Дауна — трисомия или просто внешние признаки СД — без трисомии, но опытный врач пишет Синдром Дауна. Внешие признаки вообще в расчет не беруться — если не подкреплены тремя хромосомами. уля2 пишет: тветила сделаете анализ узнаете болезнь у вас или синдром. Какие есть ТУПЫЕ врчи, просто дубы, ну возьми ты, тетя-врач, книгу прочитай, прежде, чем засорять эфир глупостями, нет лепят бред, только бедных мамочек заставляют напрасно на анализы деньги тратить и ребенка зря колоть.
Надежда : G-NA85 пишет: Болезни Дауна — нет. Есть Синдром Дауна — трисомия или просто внешние признаки СД — без трисомии, но опытный врач пишет Синдром Дауна. Внешие признаки вообще в расчет не беруться — если не подкреплены тремя хромосомами. Таня, ну это мы с Вами (и другие мамы наших деток) знаем, а многим врачам, как выразилась моя свекровь, когда я спросила, какие методики есть по обучению (она учитель): «Мне эту проблему изучать не за чем, да и некогда» Так что действительно, ДУБЫ
OlgaLD : уля2 Вопросы терминологии, не переживайте. Удачи!
клименко : Недавно я с Марусей проходила комиссию и у меня педиатр спрашивала-у ребенка синдром или болезнь Дауна?Я не стала проводить с ней всеобуч-сказала.что записано-синдром.И какую консультацию может дать такой специалист.кроме объяснения.что Дауны как растения и с ними заниматься бесполезно?
OKS : Девочки! А у нас в диагнозе просто «трисомия по 21 хромосоме». Согласитесь, звучит аккуратненько.
люся : Девочки а сейчас ведь диагнозы пишут под номерами. Синдром Дауна зашифровано выглядит вот так — Q 90. Как красиво звучит, правда?
Марийка : А у нас врачи теряются в догадках: синдром написать или болезнь. Кто хочет показаться умным в этом вопросе пишет»болезнь», а тот кто и так знает — «синдром». Кто во что горазд. Надо им уже между собой договориться. Иногда так и хочется провести специально для них ЛИКБЕЗ.
Лора : Вспомнить бы, где я читала. но суть такова, что если просто хромосомный набор и внешние признаки, то это синдром. А если еще и пороки развития, умственная отсталость, то уже болезнь. Нас тоже часто врачи спрашивают: что у вас, синдром или болезнь. И пишут везде:болезнь. Даже в заключении генетиков написано:диагноз Болезнь Дауна(транслокационный вариант). Цитирую дословно.
Лора : Вот еще нашла. Учебник для медучилищ, автор Бисярина В.П.,год издания 1981 «Детские болезни и уход за детьми». Здесь пишут о болезни Дауна. Может, поэтому все врачи так говорят? Учились по этому учебнику(или по таким же, выпущенным издательством «Медицина»).
G-NA85 : Лора пишет: . А если еще и пороки развития, умственная отсталость, то уже болезнь. А по-моему у всех наших детей есть некоторые пороки развития и УО в основном в легкой форме. Пороки тоже у всех разные, но какая-то мелочевка у каждого наскребется. Поэтому все равно правильно — СИНДРОМ ДАУНА, а не болезнь. Болезнь — это по старинке и только на территории бывшего Союза, где врачу книгу в руки взять лень. А правда чего учиться — то? Диплом есть, халат белый дали.
Meri : Синдром — определения в Интернете: Cиндром (σύνδρομον, σύνδρομο, «наравне, в согласии») - сочетание симптомов. ru.wikipedia.org/wiki/Синдром закономерное сочетание симптомов, обусловленное единым патогенезом; рассматривается как самостоятельное заболевание (например, синдром Меньера) или как стадия. www.medotvet.ru/dict/zabol/glossary/homemed_glossary_s.php закономерное сочетание признаков (симптомов), имеющих общий механизм возникновения. dic.paludarium.ru/017.htm (греч. syndrome стечение признаков болезни; син. симптомокомплекс) — совокупность симптомов, объединённых единым механизмом болезни; иногда этим термином обозначают. www.magalif.ru/index.php комплекс симптомов. www.infospid.ru/index.php состояние, развивающееся как следствие заболевания и определяющееся совокупностью клинических, лабораторных, инструментальных диагностических признаков. www.ssmu.ru/ofice/sibcem/glossary/protocols.shtml совокупность симптомов, характерных для данного заболевания. zoolife.com.ua/pageid1006.html Т.е, симптом и заболевание — в одной связке. Приходит время и все равно становится, как обзывают, синдром, болезнь — трисомия21 точнее.
Лора : OKS , о транслокации очень хорошо написано в какой-то теме Татьяной Спомер. Даже фото хромосомок. Если коротко, то это когда не 3 21-х хромосомки, а 2, но одна из них прицепилась к какой-либо еще паре. Либо они между собой срослись, образуя одну аномальную. Так и у нашего Арсения, общее кол-во хромосом 46. А у вас как написано в анализе? Или вы не делали?
OKS : Лора Мы делали. У нас то 47 к сожалению. Просто любопытно стало насчет транслокации. А написано так:кариотип: 47,ХХ, +21,21ps+. Вот. Заключение: трисомия по 21 хромосоме.
Лора : OKS почему «к сожалению»? У нас ведь тоже синдром, несмотря на 46 хромосом. Типичная внешность, порок сердца, отставание в развитие. И если с вашим диагнозом более-менее ясно- не разлепились хромосомки,то у нас не понятно, как одна затесалась в другую пару. На чаек заглянула и осталась? Как так получается, что хромосомки «убегают», интересно, есть ответ у специалистов?
lilka : Вчера вызывали на дом врача, пришла русская старушка. Мы сразу ей сказали, что у ребенка синдром Дауна. Она согласилась, сказала:»Да, у вас синдром, не сам Даун. Даун намного тяжелее». Мы хоть повеселились.
Ирэн : Нам в институте педиотрии сказали так: «Болезнь дауна — это когда поражены практически все органы, синдром дауна — малая часть».Когда мы пришли они сразу спросили «У вас болезнь или синдром?»
Ирэн : OKS Они имели в виду сопутствующие «болячки».
уля2 : ни где не могу найти что означает 13 в нашем результате анализа Кариотип: 47,ХХ,+21 может здесь кто знает?
G-NA85 : Может быть плечо 13 хромосомы легло на плечо 21 и они не разделились (это примитивное объяснение). Я спрашивала когда -то у генетика про эти плечи, хотя у Аполлинарии другой анализ. Это похоже на транслокацию. Вам нужно проконсультироваться.
уля2 : мне говорили,что похоже на траслокацию,а в заключениях написано в одном: Кариотип аномальный, несбалансированный. Трисомия по хромосоме 21. Синдром Дауна. в другом:Трисомия по хромосоме 21. уже жалею что второй раз анализ сдали, лишнюю головную боль себе нашла. Первый раз мы сдавали в нашем городе,т.к. генетический центр появился недавно была надежда,что они ошиблись, да и генетик не очень умный попался,предлагала отказаться от дочи.Вот мы и решили еще раз сдать в Новосибирске,когда ездили на операцию,там появилась эта цифра 13
Ассоль : Уля, у нас в кариотипе были цифры в квадратных скобках, но у нас мозайка, и было типа 46ХУ(15), 47ХУ(20). Насколько я поняла, число в скобках означало сколько клеток с таким набором найдено, т.к. генетик сказала, что у нас примерно 50/50. Может у вас тоже что-то подобное?
уля2 : значит у нас возможно мозаика? Наверно нужно звонить в Новосибирскую лабораторию, по мейлу не ответили,а с нашим генетиком встречаться не хочется
Светлана : уля2 пишет: Наверно нужно звонить в Новосибирскую лабораторию, по мейлу не ответили Думаю и по телефону не ответят. Это строго запрещено — врачебная тайна вообще-то не отвлеченное понятие. Они имеют право предоставлять подобную информацию только лично. Нам в свое время по телефону сказали только то, что анализ уже готов и можно подехать забрать.
Ассоль : уля2 пишет: значит у нас возможно мозаика? Насколько я поняла, у вас в кариотипе только про 47 хромосом сказано, а про 46 — ничего нет. Тогда вряд ли мозайка, при мозайке и 46 хромосом есть, и 47. Может, 13 в вашем случае — что проверено 13 клеток. А может, это еще что-то значит
G-NA85 : На мозаику не похоже, там действительно 46 хромомосом должно бытьнаписано. Узнайте у любого другого генетика, не у вашего. Нам говорили, что эта транслакационная форма, когда описывали возможные варианты. Хотя у Аполлинарии простая трисомия, а по нашим (родительским) анализам должна быть транслокация. На самом деле это без разницы все. Меня это только первые два месяца интересовало, а потом уже забила на эти хромосомы, ну их. Все равно их не видно.
Алекса и Лёша : G-NA85 А вы анализы до рождения Аполлинарии сдавали или после? Мы думаем о следующем ребенке и не знаем как поступить. Хотя после всего что с нами произошло (мы наблюдались всю беременность у генетиков: тройной тест,амниоцинтез,УЗИ — всё хорошо) слабо вериться что они нам чем-либо помогут.
Lena S. : Алекса и Лёша пишет: Хотя после всего что с нами произошло (мы наблюдались всю беременность у генетиков: тройной тест,амниоцинтез,УЗИ — всё хорошо) слабо вериться что они нам чем-либо помогут. Извините, а что, амнио не показал наличие лишней хромосомы? Или клетки не попали? Действительно, кто же может тогда что-то гарантировать.
Алекса и Лёша : Lena S. Мне только после рождения Лёши сказали, что достоверность анализа на амниоцинтез около 75% (часть околоплодной жидкости может не содержать клетки плода), и что более достоверным является исследование на 10-й неделе беременности — биопсия хориона, но там тоже достоверость 98%. И где гарантия, что не попадешь в 2%?! Причем амниоцинтез мы сделали по собственной инициативе, только потому что мне было 35 лет, врачи немного недоумевали, тройной тест хороший, сам Воеводин на УЗИ патологий не обнаружил. Причем сейчас думаю, вот что это было — предчуствие? Нет, просто хотелось все сделать правильно, как написано в книжках. Зато теперь я знаю что сделала всё возможное, чтобы не допустить данной ситуации. Ребёнок от Бога, вот всё объяснение!
Ассоль : Алекса и Лёша Получается, что по амнио вам предсказывали рождение девочки? Ведь если для анализа из околоплодных вод попали не клетки ребенка — значит, это были Ваши клетки, должен был быть набор ХХ. И по УЗИ мальчика не видели?
Алекса и Лёша : Ассоль Мы сдавали амниоцинтез для того чтобы выявить генетические патологии, а не для того чтобы узнать пол ребенка. Тема пола ребенка с генетиком не обсуждалась вообще. Мы узнали что у нас будет мальчик на 24 недели беременности, через 4 недели после амнио.
Ассоль : Алекса и Лёша пишет: Мы сдавали амниоцинтез для того чтобы выявить генетические патологии, а не для того чтобы узнать пол ребенка. Это понятно, просто пол ребенка в данном анализе определяется автоматически, когда хромосомки рассматривают. Но Вам, видимо, не сказали, а вы не спросили.
аноним : здравствуйте! подскажите пожалуйста. мы сделали анализ на кариотип и результат такой: 47,Xy,+21 и дополнительно: Q90.0 Трисомия 21, Мейотическое нерасхождение
OlgaLD : аноним Здравствуйте, добро пожаловать на форум! Это значит, что у вашего ребенка синдром Дауна, обычная форма — трисомия по 21 хромосоме. Это и обозначается кодом Q90.0. Мейотическое нерасхождение соответствует обычной трисомии. turist пишет: Кариотип 47,хх; +21, то есть во всех исследованных клетках обнаружена дополнительная 21 хромосома. И диагноз Q 90.0 (иначе говорят трисомия 21 — мейотическое нерасхождение), если написано Q 90.1, тогда Трисомия 21, мозаицизм (митотическое нерасхождение) Q 90.2 Трисомия 21, транслокация http://www.downsyndrome.borda.ru/?1-9-0-00000099-000-120-0 Транслокация и мозаицизм бывают каждый в 4-5% случаев, остальные 90%+ это обычные трисомии. Кариотип представлен 47 хромосомами вместо нормальных 46, поскольку хромосомы 21-й пары, вместо нормальных двух, представлены тремя копиями (трисомия). Отсюда цифра 47 в вашем анализе и прибавление «+21». Иными словами, «трисомия 21». Мейотическое — значит произошедшее во время мейоза, мейоз — деление клетки. Иными словами, синдром Дауна получается при нерасхождении хромосом при мейозе, делении клетки, т.е. присутствует при зачатии. Кажется, все объяснила. Добро пожаловать на форум! Почитайте эти странички: http://sunchildren.narod.ru/whatis.html http://sunchildren.narod.ru/whatdo.html http://sunchildren.narod.ru/startwith.html Пишите, откуда вы, можете найти земляков, которые помогут вам освоиться с диагнозом и найти ответы на ваши вопросы.
KaRinaSH : Народ, мне сегодня в РЦ лекцию прочитали, что типа болезнь Д и СД — это разные вещи, вот если нету порока сердца, то это СД, а у нас я прочла БД, Во как!
OlgaLD : KaRinaSH Не ты первая, не ты последняя, можешь эту тему почитать, а вообще у нас на форуме часто всплывает эта инфа от «специалистов»
Татьяна А : А у нас на бумажке с анализом написано 46, хх, der (14:21) (g10:10) +21 транслокационная
OlgaLD : Татьяна А Ну, у вас транслокационная форма, более редкая
wap.downsyndrome.borda.ru
Синдром дауна кариотипа
Клинический диагноз синдрома Дауна обычно не представляет никаких трудностей. Тем не менее для подтверждения диагноза и предоставления базы для генетического консультирования необходимо кариотипирование. Хотя различия в конкретных вариантах кариотипа, ответственных за синдром Дауна, обычно имеют небольшое влияние на фенотип пациента, они существенны для определения риска повторения.
Трисомия 21 при синдроме Дауна . Примерно у 95% всех пациентов с синдромом Дауна выявляют трисомию хромосомы 21, вызванную мейотическим нерасхождением 21 пары хромосом, как обсуждалось в предыдущей главе. Уже отмечено, что риск иметь ребенка с трисомией 21 увеличивается с возрастом матери, особенно после 30 лет. Мейотическая ошибка, ответственная за трисомию, обычно происходит в ходе материнского мейоза (около 90% случаев), преимущественно в первом делении, но около 10% случаев происходит в отцовском мейозе, обычно во втором делении.
Робертсоновская транслокация при синдроме Дауна . Около 4% пациентов с синдромом Дауна имеют 46 хромосом, одна из которых - робертсоновская транслокация между хромосомой 21q и длинным плечом одной из других акроцентрических хромосом (обычно хромосомы 14 или 22). Транслоцированная хромосома заменяет одну из нормальных акроцентрических хромосом, и кариотип пациента с робертсоновской транслокацией между хромосомами 14 и 21 — 46,XX/XY,rob(14;21)(ql0;ql0),+21.
Такая хромосома может также быть определена как der(14;21), на практике используют обе номенклатуры. В действительности пациенты с робертсоновской транслокацией, включающей хромосому 21, трисомны по генам, расположенным в длинном плече 21q.
В отличие от стандартной трисомии 21 , транслокационный синдром Дауна не показывает никакой связи с возрастом матери, но имеет сравнительно высокий риск повторения в семьях, если один из родителей, особенно мать, - носитель транслокации. По этой причине для точного генетического консультирования важно кариотипирование родителей и, возможно, других родственников.
Носители робертсоновской транслокации , включающей хромосомы 14 и 21, имеют только 45 хромосом; одна 14 и одна 21 отсутствуют и заменены транслоцированной хромосомой. Теоретически возможны шесть типов гамет, но три из них не могут привести к жизнеспособному потомству. Три типа гамет жизнеспособные, нормальные, сбалансированные и несбалансированные, имеющие как транслоцированную, так и нормальную хромосому 21. В комбинации с нормальной гаметой это может приводить к зачатию ребенка с транслокационным синдромом Дауна.
Теоретически эти три типа гамет производятся в равных количествах, таким образом, теоретический риск ребенка с синдромом Дауна должен быть 1 к 3. Тем не менее расширенные популяционные исследования показали, что несбалансированные хромосомные наборы появляются только у 10-15% потомства матерей и только у нескольких процентов потомства отцов, несущих транслокации, включающие хромосому 21.
Транслокация 21q21q при синдроме Дауна . Хромосомная транслокация 21q21q - хромосома, сформированная из двух длинных плеч хромосомы 21; бывает у нескольких процентов пациентов с синдромом Дауна. Считают, что они появляются как изохромосомы, а не робертсоновские транслокации. Большинство таких случаев возникают постзиготически, соответственно, риск повторения низкий. Тем не менее особенно важно убедиться, не является ли родитель носителем (возможно, мозаичным) данной транслокации, поскольку все гаметы носителя такой хромосомы должны также содержать 21q21q хромосому, с двойной дозой генетического материала хромосомы 21, или не иметь хромосомы 21 совсем.
Потенциальное потомство , следовательно, неизбежно имеет или синдром Дауна, или нежизнеспособную моносомию 21. Мозаичные носители имеют повышенный риск повторения, таким образом, пренатальная диагностика необходима при всех последующих беременностях.
Мозаичный синдром Дауна . Около 2% пациентов с синдромом Дауна - мозаики, обычно с популяциями нормальных клеток и с трисомией 21. Фенотип может быть мягче, чем при типичной трисомии 21. Вообще существует широкая изменчивость в фенотипах мозаичных пациентов, вероятно, отражая различные пропорции трисомных клеток у эмбриона на ранних стадиях развития. Возможно, пациенты с установленным мозаичным синдромом Дауна отражают только клинически более серьезные случаи, поскольку в легких случаях кариотипирование менее вероятно.
Частичная трисомия 21 при синдроме Дауна . Очень редко синдром Дауна диагностируют у пациентов, имеющих трисомию только по части длинного плеча хромосомы 21, и еще реже выявляют пациентов с синдромом Дауна без цитогенетически видимой хромосомной аномалии. Такие случаи представляют определенный интерес, поскольку могут указывать, какая область хромосомы 21, вероятно, ответственна за специфические компоненты фенотипа синдрома Дауна и какие области могут утраиваться, не вызывая фенотипических проявлений.
Хотя хромосома 21 содержит только несколько сотен генов, попытки согласовывать тройную дозу специфических генов со специфическими аспектами фенотипа синдрома Дауна пока имеют ограниченный успех. Наиболее примечательной стала идентификация области, критической для пороков сердца, наблюдаемых примерно у 40% пациентов с синдромом Дауна. Поиск конкретных генов, существенных для проявления фенотипа синдрома Дауна, среди случайно находящихся рядом с ними в хромосоме 21, - главная задача современных исследований, особенно на мышах в качестве модели.
Потенциально перспективное направление - исследование генно-инженерных мышей с дополнительной дозой генов из хромосомы 21 человека (или даже с полной копией хромосомы 21). Такие мыши могут проявлять фенотипические аномалии в поведении, функциях мозга и формировании сердца.
Кариотип – это совокупность признаков хромосомного набора (число, размер, форма хромосом), характерных для того или иного вида.
Исследование всех элементов кариотипа человека производится при помощи метода специальной окраски и последующего изучения хромосом в световом микроскопе. Данный метод помогает увидеть размеры и формы хромосом,  их структуру. Заболевания, сопровождающиеся патологическими изменениями кариотипа, называются хромосомными. Пример
их структуру. Заболевания, сопровождающиеся патологическими изменениями кариотипа, называются хромосомными. Пример
хромосомной болезни – синдром Дауна. Причины возникновения синдрома Дауна кроются во внутриутробном формировании хромосомной патологии плода. Так, если нормальный кариотип здорового человека состоит из 46 хромосом, то при синдроме Дауна он сформирован 47 хромосомами, у 21 пары обнаруживается добавочная хромосома, отвечающая за болезнь.
Кариотип с аномалиями обнаруживается у около 1 % всех новорожденных. Последствия таких аномалий могут колебаться от смерти до умственной отсталости и слабовыраженных аномалий. Случаи аномального числа хромосом увеличиваются с возрастом матери. Возраст будущей мамы также влияет на риск развития у ребенка синдрома Дауна: если в возрасте до 35 лет такой риск составляет 1 к 1000; то в возрасте до 40 лет риск возрастает до 1 к 214; в возрасте старше 45 лет риск увеличивается до 1 к 19.
Вариантов отклонений от нормы множество. На сегодняшний день выявлены следующие аномалии:
Происходит это уже на ранней стадии зарождения эмбриона. Одной из причин отклонений является нарушение сперматогенеза, некоторые из нарушенных сперматозоидов все же участвуют в оплодотворении яйцеклетки и, следовательно, могут являться причиной формирования эмбриона с нарушенным кариотипом.
Основным неблагоприятным фактором появления отклонений является плохая экология, провоцирующая хромосомные мутации. Все эти аномалии передаются по наследству.
На данный момент нет содержимого, классифицированного этим термином.
Мозаичная форма синдрома Дауна: как определить
Генетическая патология, вызванная изменениями в 21-й хромосоме – это мозаичный синдром Дауна. Рассмотрим его особенности, методы диагностики, лечения и профилактики.
Болезнь Дауна относится к самым распространенным врожденным генетическим расстройствам. Для нее характерно выраженное отставание в умственном развитии и ряд внутриутробных аномалий. Из-за высокой рождаемости детей с трисомией, было проведено множество исследований. Патология встречается у представителей всех народов мира, поэтому географической или расовой зависимости не установлено.
Код по МКБ-10
Эпидемиология
Согласно медицинской статистике, синдром Дауна встречается у 1 ребенка на 700-1000 родов. Эпидемиология расстройства связана с определенными факторами: наследственная предрасположенность, вредные привычки родителей и их возраст.
Закономерность распространения болезни не связана с географической, половой, национальной принадлежность или экономическим статусом семьи. Трисомия обусловлена нарушениями в процессе развития ребенка.
Причины мозаичного синдрома Дауна
Основные причины мозаичного синдрома Дауна связаны с генетическими нарушениями. У здорового человека содержится 23 пары хромосом: женский кариотип 46, ХХ, мужской 46, ХY. Одна из хромосом каждой пары, передается от матери, а вторая от отца. Болезнь развивается в результате количественного нарушения аутосом, то есть к 21-й паре присоединяется лишний генетический материал. Трисомия по 21-й хромосоме отвечает за симптомы дефекта.
Мозаичный синдром может возникать по таким причинам:
Образование аномальных гамет может быть связано с некоторыми заболеваниями половой сферы родителей, радиацией, курением и алкоголизмом, приемом медикаментов или наркотических веществ, а также с экологической обстановкой места проживания.
Около 94% синдрома связано с простой трисомией, то есть: кариотип 47, ХХ, 21+ или 47, ХY, 21+. Копии 21-й хромосомы есть во всех клетках, так как во время мейоза в родительских клетках нарушено деление парных хромосом. Около 1-2% случаев обусловлены нарушением митоза клеток зародыша на стадии гаструлы или бластулы. Мозаицизм характеризуется трисомией в дериватах пораженной клетки, в то время как остальные имеют нормальный хромосомный набор.
При транслокационной форме, которая встречается у 4-5% пациентов, 21-я хромосома или ее фрагмент транслоцируется к аутосоме при мейозе, проникая с ней в только что сформированную клетку. Основными объектами транслокации выступают 14, 15, реже 4, 5, 13 или 22 хромосомы. Подобного рода изменения могут быть случайными или наследоваться от родителя, который выступает носителем транслокации и нормального фенотипа. Если подобные нарушения есть у отца, то риск рождения больного ребенка составляет 3%. При носительстве со стороны матери – 10-15%.
Факторы риска
Трисомия является генетическим заболеванием, которое нельзя приобрести в течение жизни. Факторы риска его развития не связаны с образом жизни или этнической принадлежностью. Но шансы родить больного ребенка увеличиваются при таких обстоятельствах:
Существуют предположения, что развитие недомогания может быть связано с возрастом, в котором бабушка родила мать и другими факторами. Благодаря преимплантационной диагностике и другим исследовательским методам, риск рождения ребенка Дауна существенно снижается.
Развитие генетического заболевания связано с хромосомной аномалией, при которой у больного вместо 46 появляется 47 хромосом. Патогенез мозаичного синдрома имеет иной механизм развития. Половые клетки-гаметы родителей имеют нормальное количество хромосом. Их слияние привело к формированию зиготы с кариотипом 46, ХХ или 46, XY. В процессе деления первоначальной клетки ДНК произошел сбой, и распределение было неправильным. То есть часть клеток получила нормальный кариотип, а часть – патологический.
Подобного рода аномалия встречается в 3-5% случаев болезни. Она имеет положительный прогноз, поскольку здоровые клетки частично компенсируют генетическое нарушение. Такие дети рождаются с внешними признаками синдрома и отставанием в развитии, но их выживаемость значительно выше. У них реже встречаются внутренние патологии, несовместимые с жизнью.
Симптомы мозаичного синдрома Дауна
Аномальная генетическая особенность организма, возникающая при увеличении числа хромосом, имеет ряд внешних и внутренних признаков. Симптомы мозаичного синдрома Дауна проявляются отставанием в умственном и физическом развитии.
Основные физические симптомы болезни:
Заболевание вызывает ряд отклонений в развитии и здоровье. Прежде всего, это когнитивная отсталость, пороки сердца, проблемы с зубами, глазами, спиной, слухом. Склонность к частым инфекционным и респираторным заболеваниям. Степень проявлений болезни зависит от врожденных факторов и правильно подобранного лечения. Большинство малышей обучаемы, несмотря на умственное, физическое и психическое отставание.
Первые признаки
Мозаичный синдром Дауна имеет менее выраженные симптомы, в отличие от классической формы расстройства. Первые признаки можно увидеть на УЗИ на 8-12 неделе беременности. Они проявляются увеличением воротниковой зоны. Но ультразвуковое исследование не дает 100% гарантии наличия болезни, а позволяет оценить вероятность пороков развития у плода.
Наиболее характерны внешние симптомы, с их помощью врачи предположительно диагностируют патологию сразу после рождения малыша. Для дефекта характерно:
При дальнейшем обследовании выявляют такие проблемы:
Кроме внешних симптомов, синдром имеет и внутренние нарушения:
Вышеописанная симптоматика требует постоянного лечения для поддержания нормального состояния организма. Именно врожденные пороки являются причиной короткой жизни даунов.
Внешние признаки мозаичной формы синдрома Дауна
В большинстве случаев внешние признаки мозаичной формы синдрома Дауна проявляются сразу после рождения. Из-за высокой распространенности генной патологии, ее симптомы детально изучены и описаны.
Изменения в 21-й хромосоме характеризуются такими внешними признаками:
- Аномальное строение черепа.
- Врожденные дефекты ротовой полости.
- Добавочные кожные складки.
- Патологии развития опорно-двигательного аппарата
- Деформация грудной клетки.
Это наиболее заметный и выраженный симптом. В норме у малышей голова больше, чем у взрослых. Поэтому любые деформации видны сразу после рождения. Изменения касаются строения черепной коробки и лицевого черепа. У больного диспропорция в области костей темени. Также наблюдается уплощение затылка, плоское лицо и выраженный глазной гипертелоризм.
Человек с данным заболеванием напоминает представителя монголоидной расы. Подобные изменения проявляются сразу после рождения и сохраняются на протяжении всей жизни. Кроме этого стоит отменить косоглазие у 30% больных, наличие кожной складки у внутреннего угла века и пигментирование радужной оболочки глаза.
Подобного рода расстройства диагностируют у 60% больных. Они создают трудности при кормлении ребенка, замедляя его рост. У человека с синдромом изменена поверхность языка из-за утолщенного сосочкового слоя (бороздчатый язык). В 50% случаев встречается готическое небо и нарушения сосательного рефлекса, полуоткрытый рот (мышечная гипотония). В редких случаях наблюдаются аномалии типа «волчья пасть» или «заячья губа».
Данное нарушение встречается в 40% случаев. Недоразвитые хрящи формируют неправильную ушную раковину. Ушки могут быть оттопырены в разные стороны или располагаться ниже уровня глаз. Несмотря на то, что дефекты являются косметическими, они могут вызывать серьезные проблемы со слухом.
Встречаются у 60-70% больных. Каждая кожная складка вызвана недоразвитием костей и их неправильной формой (кожа не натягивается). Данный внешний признак трисомии проявляется как избыток кожи на шее, утолщения в локтевом суставе и поперечная складка на ладони.
Возникают из-за нарушения внутриутробного развития плода. Соединительная ткань суставов и некоторые кости не успевают полностью сформироваться до рождения. Самые распространенные аномалии: короткая шея, повышенная подвижность суставов, короткие конечности и деформированные пальцы.
Данная проблема связана с недоразвитостью костной ткани. У больных наблюдается деформация грудного отдела позвоночника и ребер. Чаще всего диагностируют выпирающую грудину над поверхностью грудной клетки, то есть килевидную форму и деформацию, при которой в области солнечного сплетения есть воронкообразное углубление. Оба нарушения сохраняются по мере взросления и роста. Они провоцируют нарушения в строении дыхательного аппарата и сердечно-сосудистой системы. Такие внешние симптомы указывают на плохой прогноз болезни.
Главная особенность мозаичной формы синдрома Дауна в том, что при ней многие вышеописанные симптомы могут отсутствовать. Это усложняет дифференциацию патологии с другими хромосомными аномалиями.
Синдром имеет несколько видов, рассмотрим их:
Осложнения и последствия
Хромосомный мозаицизм вызывает последствия и осложнения, которые негативно сказываются на состоянии здоровья и существенно ухудшают прогноз заболевания.
Рассмотрим основные опасности трисомии:
Кроме вышеописанных осложнений, трисомия характеризуется проблемами с щитовидной железой, слабостью костей, плохим зрением, потерей слуха, ранней менопаузой и непроходимостью кишечника.
Диагностика мозаичного синдрома Дауна
Выявить генетическую патологию можно еще до рождения. Диагностика мозаичного синдрома Дауна основана на исследовании кариотипа крови и клеток ткани. На ранних сроках беременности проводится биопсия хориона, позволяющая выявить признаки мозаицизма. Согласно статистике только 15% женщин, узнавших о генетических аномалиях у ребенка, решаются его оставить. В остальных случаях показано преждевременно прерывание беременности – аборт.
Рассмотрим наиболее достоверные методы диагностики трисомии:
Вышеописанные исследования позволяют оценить риск рождения ребенка с синдромом, но они не дают абсолютной гарантии. При этом процент ошибочных результатов диагностики, проводимой во время беременности невелик.
Диагностика геномной патологии начинается в период вынашивания. Анализы проводятся на ранних сроках беременности. Все исследования на наличие трисомии называются скринингами или отсевами. Их сомнительные результаты позволяют заподозрить наличие мозаицизма.
- Второй триместр – анализы на ХГЧ и эстриол, АФП и ингибин-А. В отдельных случаях проводят исследование генетического материала. Для его забора делают прокол матки через живот.
- Синдром Шерешевского-Тернера
- Синдром Эдвардса
- Синдром де Ля Шапеля
- Врожденный гипотиреоз
- Другие формы хромосомных аномалий
- Массажи – мышечная система, как младенца, так и взрослого с данным синдромом недостаточно развита. Специальная гимнастика способствует восстановлению тонуса мышц и поддерживает их в нормальном состоянии. Особое внимание уделяется гидромассажам. Плаванье и водная гимнастика улучшают моторику, укрепляют мышцы. Популярностью пользуется дельфинотерапия, когда больной плавает с дельфинами.
- Консультации диетолога – пациенты с трисомией имеют проблемы с лишним весом. Ожирение может спровоцировать различные нарушения, самыми частыми выступают расстройства сердечно-сосудистой системы и пищеварительного тракта. Диетолог дает рекомендации по питанию и при необходимости назначает диету.
- Консультации логопеда – для мозаицизма, как и для других видов синдрома, характерны нарушения в развитии речи. Занятия с логопедом помогут больному правильно и четко выражать свои мысли.
- Специальная программа обучения – дети с синдромом отстают в развитии от сверстников, но они обучаемы. При правильном подходе, ребенок может освоить базовые знания и навыки.
- Своевременное лечение любых заболеваний и здоровый образ жизни. Повышенная активность улучшает кровообращение, защищая яйцеклетки от кислородного голодания.
- Правильное питание и нормальный вес. Витамины, минералы и другие питательные вещества не только укрепляют иммунитет, но и поддерживают гормональное равновесие. Лишний вес или чрезмерная худоба нарушают гормональный баланс, и провоцируют сбои в созревании и развитии половых клеток.
- Подготовка к беременности. За пару месяцев до планируемого зачатия необходимо пройти консультацию у гинеколога и начать принимать витаминно-минеральные комплексы. Особое внимание стоит уделить фолиевой кислоте, витаминам В и Е. Они нормализуют работу половых органов и улучшают обменные процессы в половых клетках. Не стоит забывать, что риск родить ребенка с отклонениями возрастает у пар, где возраст будущей матери больше 35 лет, а отца больше 45.
- Дородовая диагностика. Анализы, скрининги и ряд других диагностических процедур, проводимых во время беременности, позволяют выявить серьезные нарушения у плода и принять решение о дальнейшем вынашивании или абортировании.
- Джейми Брюэр – актриса, известная по роли в сериале «Американская история ужасов». Девушка не только снимается в кино, она еще и модель. Джейми участвовала в показе Mercedes-Benz Fashion Week в Нью-Йорке.
- Раймонд Ху – молодой художник из Калифорнии, США. Особенность его картин в том, что он рисует их по старинной китайской методике: на рисовой бумаге, акварелью и тушью. Самые популярные работы парня – это портреты животных.
- Паскаль Дюкенн – актер, обладатель серебряной премии Каннского кинофестиваля. Прославился благодаря роли в фильме Жако ванн Дормеля «День восьмой».
- Рональд Дженкинс – всемирно известный композитор и музыкант. Его любовь к музыке началась с подарка – синтезатора, полученного в детстве на Рождество. На сегодняшний день Рональд по праву считается гением электронной музыки.
- Карен Гафнии – ассистент преподавателя, спортсменка. Девушка занимается плаваньем и участвовала в марафоне на Ла-Манше. Она стала первым человеком с мозаицизмом, который проплыл 15 км при температуре воды +15°C. У Карен есть свой благотворительный фонд, который представляет интересы людей с хромосомными патологиями.
- Тим Харрис – ресторатор, владелец «самого дружелюбного ресторана в мире». Кроме вкусного меню, заведение Тима предлагает бесплатные объятия.
- Мигель Томасин – участник группы Reynols, барабанщик, гуру экспериментальной музыки. Парень исполняет как свои песни, так и каверы известных рок-музыкантов. Занимается благотворительностью, выступает в центрах и на концертах для поддержки больных детей.
- Богдан Кравчук – первый в Украине человек с синдромом Дауна, поступивший в университет. Парень живет в Луцке, увлекается наукой, имеет много друзей. Богдан поступил в Восточноевропейский национальный университет имени Леси Украинки на исторический факультет.
Если по результатам анализов установлен высокий риск трисомии, то беременной назначают консультацию генетика.
Инструментальная диагностика
Для выявления внутриутробных патологий у плода, в том числе и мозаицизма, показана инструментальная диагностика. При подозрении на синдром Дауна проводят скрининги в течение всей беременности, а также ультразвуковое исследование для измерения толщины задней части шейки плода.
Самым опасным методом инструментальной диагностики является амниоцентез. Это исследование околоплодных вод, которое проводят на сроке от 18 недель (необходим достаточный объем жидкости). Основанная опасность данного анализа в том, что он может привести к инфицированию плода и матери, разрыву плодного пузыря и даже выкидышу.
Дифференциальная диагностика
Мозаичная форма изменений в 21-й хромосоме требует тщательного исследования. Дифференциальная диагностика синдрома Дауна проводится с такими патологиями:
В некоторых случаях мозаицизм по половым хромосомам XX/XY приводит к истинному гермафродитизму. Дифференциация необходима и для мозаицизма гонад, которые выступают частным случаем органной патологии, возникающей на поздних стадиях эмбрионального развития.
К кому обратиться?
Лечение мозаичного синдрома Дауна
Терапия хромосомных заболеваний невозможна. Лечение мозаичного синдрома Дауна пожизненное. Оно направлено на устранение пороков развития и сопутствующих заболеваний. Человек с данным диагнозом находится под контролем таких специалистов: педиатр, психолог, кардиолог, психиатр, эндокринолог, окулист, гастроэнтеролог и другие. Все лечение направлено на социально-семейную адаптацию. Задача родителей обучить малыша полному самообслуживанию и контакту с окружающими.
Лечение и реабилитация даунов состоит из таких процедур:
Больным показана общеукрепляющая терапия, очень часто назначают психостимуляторы, нейрометаболические и гормональные препараты. Также необходим регулярный прием витаминов. Всю медикаментозную терапию комбинируют с лечебно-педагогической коррекцией. Врожденные патологии и сложные заболевания требуют хирургического вмешательства.
Профилактика
На сегодняшний день не существует надежных методов предупреждения генетических заболеваний. Профилактика мозаичного синдрома Дауна состоит из таких рекомендаций:
Но даже выполнение всех профилактических мероприятий не может дать 100% гарантии рождения полностью здорового малыша. Трисомия – это случайная генетическая аномалия, от которой не застрахована ни одна женщина.
Мозаичный синдром Дауна имеет более положительный исход, в отличие от классической формы патологии. Прогноз обусловлен тем, что здоровые клетки частично компенсируют генетический дефект. Но ребенок все равно будет иметь внешние признаки трисомии и характерное для нее отставание в развитии. Но выживаемость таких больных намного выше, у них реже встречаются пороки развития, которые несовместимы с жизнью.
Известные люди с мозаичной формой синдромом Дауна
Изменения в 21-й хромосоме приводят к необратимым последствиям, которые не поддаются лечению. Но, несмотря на это, среди родившихся с трисомией есть художники, музыканты, писатели, актеры и множество других состоявшихся личностей. Известные люди с мозаичной формой синдрома Дауна смело заявляют о своей болезни. Они яркий пример того, что при желании можно справиться с любой проблемой. Геномное нарушение есть у таких знаменитостей:







Как показывает практика и реальные примеры, несмотря на все осложнения и проблемы генной патологии, при правильном подходе к ее коррекции, можно вырастить успешного и талантливого ребенка.
Человеческий геном состоит из 46 хромосом, объединенных в 23 пары. Из них 44 являются соматическими, то есть отвечают за строение и признаки всего тела человека. И только одна пара хромосом несет информацию о его половой принадлежности и обуславливает различия мужчин и женщин.
Обе половые хромосомы женщин одинаковы по строению и обозначаются в генетике буквой X. А у мужчин эта пара представлена разными хромосомами – X и Y.
Синдром Клайнфельтера: кариотип
Синдромом Клайнфельтера считают такое изменение хромосомного набора, при котором к кариотипу XY прибавляется одна или несколько X хромосом. Соответственно и страдают от этого заболевания только носители Y хромосомы, то есть мужчины.
Человек с синдромом Клайнфельтера имеет хромосомный набор, отличающийся от нормы всего лишь одной парой хромосом — как раз той, которая отвечает за половые признаки.
Для наглядности мы постарались изобразить кариотип больного с синдромом Клайнфельтера на рисунке:
Разнообразие вариантов
Синдром Клайнфельтера может быть представлен разными цитогенетическими вариантами, которые определяют также различие в выраженности симптомов и тактике ведения больных.

Истоки болезни
 Причины появления синдрома Клайнфельтера кроются в нерасхождении хромосом при делении клетки.
Причины появления синдрома Клайнфельтера кроются в нерасхождении хромосом при делении клетки.
Согласно статистике треть больных получают лишнюю хромосому от отцовского сперматозоида, а другие две трети – от материнской яйцеклетки.
Факторами риска для появления этого заболевания традиционно считают вирусные инфекции, нарушения в работе иммунных систем родителей и поздний возраст матери.
Постановка диагноза
При врач может опираться на уровень гормонов в анализе крови, результаты спермограммы, УЗИ мошонки и биопсии яичек. Но окончательно подтвердить диагноз можно только по итогам анализа крови на кариотип, свойственный синдрому Клайнфельтера.
Для этого выделенные из крови лейкоциты помещают в питательную среду и затем исследуют на наличие хромосомной аномалии в их ДНК.
Современный анализ крови позволяет точно дифференцировать любое генетическое заболевание и со 100%-й вероятностью отличить, например, синдром от синдрома еще на этапе беременности. Для этого проводят забор клеток эмбриона или околоплодных вод.
В развитых странах многие хромосомные аномалии, включая синдром Клайнфельтера, выявляются еще во время беременности, поскольку женщины, планирующие материнство в позднем возрасте, стараются максимально исключить риск рождения больного малыша.

В США при наличии такой аномалии у будущего ребенка около половины женщин предпочитают прервать беременность. В России анализ на кариотипирование не входит в широкую практику, он проводится, только если по результатам скрининга беременной появляются подозрения о наличии генетических отклонений у плода.
Во многих случаях синдром выявляется намного позже – при возникновении характерных признаков в период взросления.
Несмотря на успехи современной медицины, примерно половина случаев синдрома Клайнфельтера остаются вообще нераспознанными, хотя больные и обращаются к врачам с жалобами на увеличение молочных желез, нарушение эрекции и бесплодие.